En el campo de la química y la ciencia de materiales, la capacidad para predecir propiedades, resultados de reacciones y comportamiento de materiales es fundamental para acelerar el desarrollo y la innovación. Tradicionalmente, estas predicciones se han basado en modelos de aprendizaje automático convencionales que requieren una featurización rigurosa, es decir, la conversión de datos químicos complejos en vectores numéricos específicos, lo que demanda un conocimiento especializado y una preparación exhaustiva de datos. Sin embargo, la llegada de los modelos de lenguaje a gran escala (LLMs, por sus siglas en inglés) afinados está marcando un punto de inflexión, abriendo nuevas posibilidades para investigadores y profesionales sin necesidad de manejar complejos procesos tradicionales de extracción de características. Los LLMs están entrenados inicialmente con volúmenes masivos de texto de diversas fuentes, permitiéndoles comprender y generar lenguaje natural con gran precisión. Sin embargo, su conocimiento químico específico suele ser limitado en el entrenamiento general.
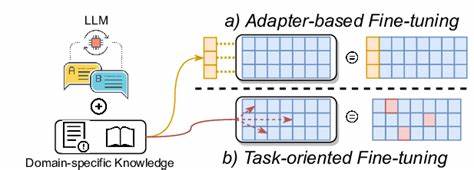
Afinar estos modelos con conjuntos de datos específicos de química y ciencia de materiales les proporciona un conocimiento enfocado y mejora su capacidad predictiva. Esta técnica, conocida como fine-tuning, adapta parcialmente los parámetros del modelo para que pueda responder a problemas específicos como clasificaciones binarias de propiedades o predicciones detalladas. Una de las grandes ventajas del enfoque basado en LLMs afinados es la capacidad para trabajar directamente con representaciones textuales comunes en química, tales como el formato SMILES, nombres IUPAC, o incluso cadenas de aminoácidos en biología molecular. Esto elimina la necesidad de transformaciones complejas y facilita el acceso a estas herramientas para científicos que no son expertos en aprendizaje automático. Diversos estudios recientes han demostrado que los LLMs afinados pueden superar o igualar el desempeño de métodos tradicionales en tareas concretas.
Por ejemplo, predicciones de energías adhesivas en polímeros modelados mediante cadenas de copolímeros, clasificación de propiedades termodinámicas de monómeros, o estimación de puntos de fusión en moléculas pequeñas, han sido abordados con éxito usando modelos como GPT-J-6B, Llama-3.1-8B y Mistral-7B. En estos escenarios, la precisión alcanzada en clasificaciones binarias suele superar el 75%, en ciertos casos llegando hasta un 96%, lo que representa una mejora clara sobre modelos convencionales como random forest o XGBoost. El uso de LLMs para predecir resultados de reacciones químicas es otra área prometedora. La complejidad inherente y la especificidad de las reacciones dificultan el desarrollo de teorías generales, pero los modelos afinados han logrado clasificar la energía de activación o el rendimiento de catalizadores con gran eficacia.
Incluso con conjuntos de datos experimentales limitados, la afinación cuidadosa y el uso de representaciones textuales adecuadas han permitido alcanzar tasas de acierto superiores al 80% en problemas que tradicionalmente requerían extensos recursos computacionales y tiempo. Además, las aplicaciones prácticas de estos modelos se extienden a sistemas y tecnologías químicas complejas, como la predicción de la capacidad de adsorción de gases en estructuras como metal-organic frameworks (MOFs), la clasificación de materiales para almacenamiento de hidrógeno, o la optimización de procesos de desalinización térmica. En todos estos contextos, los modelos afinados han mostrado capacidad para manejar múltiples variables en un solo prompt, combinando parámetros numéricos y textuales, y generando predicciones confiables que pueden acelerar el diseño experimental y la toma de decisiones. Un aspecto fundamental es la capacidad de estos modelos para aprender con datasets relativamente pequeños, lo que es crucial en química debido a que a menudo la cantidad de datos disponibles es limitada. Resultados obtenidos con conjuntos de datos tan reducidos como 20 o 30 ejemplos han podido ser mejorados mediante ajustes en los hiperparámetros y aumentando las épocas de entrenamiento, alcanzando niveles aceptables de predictividad, una ventaja significativa frente a otros métodos que requieren grandes cantidades de datos para obtener resultados óptimos.
El tratamiento directo de datos textuales facilita además la interpretación y comunicación entre especialistas de distintas áreas, fomentando la colaboración interdisciplinaria. Los prompts usados para entrenar los modelos pueden incluir preguntas formuladas de manera natural, seguidas de respuestas binarias comprensibles para cualquier investigador, lo que también contribuye a la democratización del uso de inteligencia artificial en la química. Sin embargo, existen desafíos importantes. Uno de los más evidentes es el desequilibrio inherente en muchos conjuntos de datos reales, donde las muestras que muestran resultados exitosos o propiedades óptimas son minoría. Hacer frente a esta desproporción requiere estrategias específicas durante la afinación para evitar modelos sesgados o poco confiables.
Además, a medida que aumenta la complejidad de los sistemas y las clases de salida (como en predicciones multicategoría o modelos de regresión), se observa que se requieren más datos y cuidados en la preparación y ajuste del modelo. A pesar de estas limitaciones, la adaptación de LLMs representa una herramienta poderosa y versátil para la química actual y futura. Su implementación puede reducir significativamente el tiempo y los recursos necesarios para evaluar compuestos, optimizar rutas sintéticas o diseñar nuevos materiales. Esta metodología también puede complementar técnicas computacionales avanzadas, como cálculos de química cuántica o simulaciones moleculares, sirviendo como filtro previo para seleccionar candidatos prometedores. En resumen, la evaluación de modelos de lenguaje a gran escala afinados muestra un gran potencial para aplicaciones químicas reales.
Ofrecen una plataforma accesible que combina el conocimiento textual generalista con datos específicos del área, permitiendo predecir propiedades, resultados y comportamientos con una precisión sorprendente y con una curva de aprendizaje baja para el usuario. Esta innovación fomenta la eficiencia y creatividad en la investigación química, al tiempo que impulsa una mayor integración de la inteligencia artificial en la ciencia de materiales y la experimentación química de vanguardia.