La historia evolutiva de los seres humanos ha sido, durante mucho tiempo, un campo fascinante y complejo, donde las evidencias fósiles, arqueológicas y genéticas convergen para construir un relato sobre nuestros orígenes. Sin embargo, la evolución humana no es una línea simple y directa, sino una trama intricada de poblaciones que se separaron, mezclaron y experimentaron variaciones en tamaños poblacionales a lo largo del tiempo. Recientemente, un avance científico ha arrojado luz sobre una estructura ancestral muy profunda que comparten todas las poblaciones humanas modernas, revelada mediante un innovador modelo llamado "cobraa", basado en la teoría de la cóalescencia estructurada. Este enfoque redefine lo que se sabe sobre nuestra ascendencia común y pone en cuestión algunos supuestos previos sobre la historia humana. La cóalescencia, en genética poblacional, es un concepto que describe cómo los linajes genéticos actuales se unen en ancestros comunes a lo largo del tiempo.
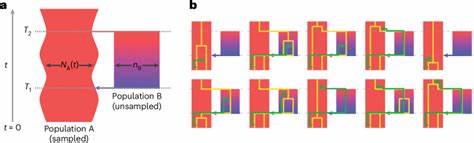
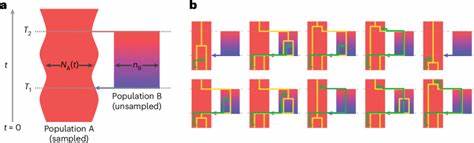
Tradicionalmente, se ha utilizado la cóalescencia bajo el supuesto de que la población ancestral fue panmítica, es decir, con reproducción aleatoria sin estructura interna. Sin embargo, esta hipótesis resulta insuficiente para explicar ciertos patrones observados en los genomas humanos modernos, como señales de admixtura antigua o evidencias de eventos demográficos complejos. Para superar estas limitaciones, los investigadores desarrollaron el modelo cobraa, que permite representar explícitamente la posibilidad de que la población ancestral estuviera estructurada en dos grupos que se separaron hace aproximadamente 1.5 millones de años y que volvieron a mezclarse hace unos 300,000 años. Este modelo aplica una técnica avanzada que extiende el marco del cóalescencia secuencialmente markoviana, incorporando no solo el tiempo hacia el ancestro común de dos linajes, sino también su desplazamiento a través de poblaciones estructuradas.
Utilizando secuencias genómicas completas y de alta cobertura de múltiples individuos de diversas poblaciones humanas, cobraa pudo identificar que el genoma moderno contiene una mezcla de dos linajes ancestrales en una proporción aproximada del 80% y 20%, con la evidencia clara de un evento de mezcla puntual en el pasado remoto. Uno de los hallazgos más significativos de este estudio fue la detección de un cuello de botella pronunciado en la población mayoritaria poco después de la separación de las dos poblaciones ancestrales, lo cual implica que la población predominante experimentó una reducción drástica en el tamaño antes de la mezcla. Esta reducción puede estar asociada con eventos ecológicos o demográficos que favorecieron cierta rama genealogía, impactando profundamente la diversidad genética que observamos en el presente. Otro aspecto fascinante es la selección negativa evidenciada contra material genético procedente de la población minoritaria en el genoma actual. Se observó que las regiones derivadas del linaje minoritario tenían mayor correlación con la proximidad a secuencias codificantes, un indicio de que esos segmentos serían potencialmente deletéreos cuando se introdujeron en el contexto genético de la población mayoritaria.
Lo que sugiere una purga activa de material menos adaptado a través de la selección natural posterior al evento de admixtura. Además, el análisis reveló una fuerte asociación entre las regiones del genoma asignadas a la población mayoritaria y la divergencia genética con los homínidos arcaicos como los neandertales y los denisovanos. Esto sugiere que la población mayoritaria representaba el linaje ancestral común que dio lugar no solo a los humanos modernos sino también a estas especies cercanas, confirmando la interconexión y la compleja naturaleza de la evolución humana en el Pleistoceno. El desarrollo del modelo cobraa representa un avance conceptual y metodológico en la inferencia genética. A diferencia de métodos previos, como PSMC, que asumen panmixia, cobraa incorpora explícitamente la estructura poblacional y puede discriminar entre cambios en tamaño efectivo y la estructura demográfica.
El modelo utiliza información adicional en las transiciones condicionadas entre tiempos de coalescencia adyacentes, lo que le otorga capacidad para identificar eventos de admixtura y separaciones más antiguas que los enfoques previos no lograban discernir. Los investigadores aplicaron cobraa a datos genómicos obtenidos de proyectos internacionales como el 1000 Genomes Project y el Human Genome Diversity Project, abarcando una amplia diversidad poblacional global. Los resultados mostraron consistencia en el patrón de estructura profunda con parámetros similares para las poblaciones estudiadas, reforzando la idea de que este evento ancestral está presente en todos los humanos modernos, independientemente de su origen geográfico. Esto también fue corroborado mediante simulaciones que replicaban el patrón genómico observado. En cuanto a la aplicación práctica de cobraa, también se desarrolló un enfoque extendido denominado cobraa-path, que permite inferir las regiones específicas del genoma actual que provienen de cada una de las poblaciones ancestrales.
Esta capacidad es fundamental para estudiar los efectos funcionales y evolutivos de la admixtura, incluyendo la identificación de regiones enriquecidas o empobrecidas en material de un linaje particular, y para analizar cómo estas regiones se relacionan con funciones biológicas específicas. La investigación encontró que los genes asociados con regiones enriquecidas en material del linaje minoritario están sobrerrepresentados en funciones neuronales y de procesamiento neuronal, mientras que las regiones empobrecidas presentan una diversidad de funciones biológicas y muestran señales de selección negativa. Estos patrones advierten que la admixtura antigua pudo contribuir a la diversidad funcional, pero también que la selección regularizó el genoma influenciada por las condiciones evolutivas posteriores. En cuanto a la reconstrucción evolutiva más amplia, el estudio sitúa el evento de separación poblacional profunda en un contexto temporal cercano a la aparición o divergencia de especies del género Homo que coexistieron en África durante el Pleistoceno. La mezcla aproximadamente hace 300 mil años podría coincidir con fases tempranas del origen de Homo sapiens, apoyando modelos que sugieren una historia compleja y reticulada de poblaciones en lugar de un origen único y simple.
Este trabajo también tiene implicaciones para la comprensión de la relación entre el ancestro común de los humanos modernos y los homínidos arcaicos, dado que el linaje mayoritario parece estar directamente relacionado con neandertales y denisovanos, mientras que el minoritario representa una rama menos involucrada en la divergenica común con estos grupos. Más allá de la especie humana, el modelo cobraa fue aplicado a genomas de otras especies mamíferas, como el delfín común y ciertos simios, con resultados variables. Esta comparativa destaca que mientras algunos animales muestran evidencias de eventos análogos de estructura población y admixtura, otros no presentan señales significativas, enfatizando que cada historia evolutiva es particular y puede ser explorada con esta herramienta. Es importante mencionar que el modelo, aunque potente y novedoso, presenta algunas limitaciones. La complejidad del pasaje demográfico humano puede involucrar múltiples eventos de división y contacto, migraciones continuas y selección purificadora, aspectos que no son completamente modelados en la versión actual de cobraa.
Tampoco la heterogeneidad en tasas de mutación y recombinación o la influencia de selección natural en el genoma son consideradas completamente, aunque la robustez demostrada en simulaciones sugiere que las conclusiones son confiables. Finalmente, el hallazgo de esta estructura ancestral profunda redefine la narrativa genética humana tradicional y abre nuevos horizontes para la investigación. Es probable que en el futuro, combinando este enfoque con otros datos genómicos, arqueológicos y paleontológicos se pueda dilucidar con mayor precisión la complejidad de los procesos que llevaron a la formación de nuestra especie y a la diversidad genética actual. Los avances en secuenciación y análisis, optimizados por métodos como cobraa, permiten ahora explorar eventos evolutivos de millones de años atrás con gran detalle, reescribiendo la historia común que los seres humanos compartimos y cómo evolucionaron nuestras poblaciones ancestrales.