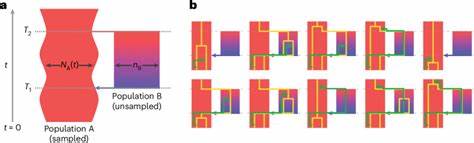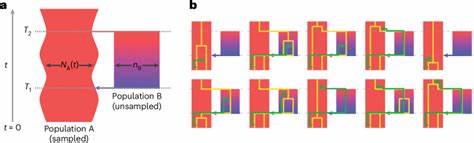
La comprensión del origen y la evolución del ser humano ha sido durante mucho tiempo un tema de fascinación y estudio profundo en los campos de la genética, la antropología y la biología evolutiva. Los avances recientes en el análisis genómico han permitido desvelar detalles complejos sobre cómo diferentes poblaciones ancestrales se separaron y luego se volvieron a reunir a lo largo de millones de años, moldeando la diversidad genética que observamos hoy en día en todas las poblaciones humanas. Recientemente, un equipo de investigadores ha desarrollado un modelo coalescente estructurado llamado cobraa, que emplea un enfoque basado en un modelo oculto de Markov para analizar datos genómicos y reconstruir con mayor precisión la historia ancestral de los humanos. Este modelo representa explícitamente un escenario donde una población ancestral se divide en dos grupos que posteriormente se vuelven a unir a través de un evento de mezcla, o admixtura, en el pasado distante. Las investigaciones utilizando el modelo cobraa revelan una estructura ancestral profunda compartida por toda la humanidad moderna.
Según los resultados, hace aproximadamente 1.5 millones de años existieron dos poblaciones ancestrales que divergieron claramente y permanecieron aisladas durante cientos de miles de años. Posteriormente, alrededor de 300,000 años atrás, se produjo un evento de admixtura entre estas dos poblaciones, donde la mezcla genética no fue equitativa, sino que aproximadamente el 80% de nuestro genoma moderno derivaría de una población mayoritaria y el 20% restante de una población minoritaria. Este descubrimiento es fundamental porque aporta una nueva capa de evidencia sobre la compleja historia evolutiva humana, extendiendo nuestra comprensión más allá del modelo clásico de una única población panmíctica o mezclada indefinidamente. La existencia de esta estructura ancestral sugiere que durante un prolongado periodo en la historia evolutiva, estas poblaciones estuvieron separadas geográfica o reproductivamente, acumulando diferencias genéticas significativas, para luego volver a mezclarse configurando la base genética común de los humanos actuales.
El modelo coalescente utilizado se basa en la teoría del proceso coalescente, un marco probabilístico que traza hacia atrás en el tiempo las genealogías de alelos en una muestra genética, permitiendo inferir eventos demográficos como cambios en el tamaño poblacional y eventos de admixtura. Sin embargo, la gran innovación del modelo cobraa reside en su capacidad para incorporar explícitamente la estructura poblacional y eventos de separación y remezcla, superando las limitaciones de modelos anteriores que asumían una población sin subestructura. Los investigadores aplicaron este modelo a datos genómicos de alta cobertura de distintas poblaciones humanas provenientes de proyectos como el 1000 Genomes Project y el Human Genome Diversity Project. Las inferencias apoyan la existencia de un fuerte cuello de botella en la población mayoritaria justo después de la divergencia, seguido de un incremento progresivo en el tamaño efectivo poblacional hasta la fase de admixtura. El análisis a nivel genómico también permitió identificar regiones específicas que derivan de cada una de las poblaciones ancestrales.
Resulta especialmente interesante que el material genético proveniente de la población minoritaria presenta una correlación significativa con la distancia a las secuencias codificantes, indicando que estos segmentos probablemente fueron deletéreos o tuvieron menores tasas de adaptación en el fondo genético de la población mayoritaria, lo que apunta a la acción selectiva negativa post-admixtura. Adicionalmente, al comparar regiones del genoma atribuibles a la población mayoritaria con la divergencia genética hacia los genomas de Neandertales y Denisovanos, se identificó una fuerte asociación. Esto sugiere que la población mayoritaria pudo ser también la principal antecesora de estos homínidos arcaicos, reforzando la hipótesis de que el origen común con estos grupos se encuentra en esta rama ancestral dominante. Estos hallazgos integran información fósil y genética, coincidiendo con fechas estimadas para la aparición de Homo sapiens anatómicamente modernos y proponiendo un modelo donde la evolución humana temprana se caracterizó por un aislamiento prolongado entre subpoblaciones y posteriores eventos de admixtura que engendraron la diversidad genética contemporánea. Este modelo aporta un marco para comprender y reinterpretar eventos clave en la historia humana, como los flujos de genes conocidos desde Neandertales y Denisovanos hacia humanos modernos fuera de África, y las evidencias más recientes de contribuciones genéticas de poblaciones arcaicas desconocidas dentro de África.
Más allá de la importancia para el entendimiento del pasado humano, la metodología propuesta tiene un impacto significativo en la forma en que se analizan los datos genómicos, permitiendo mejorar la precisión en la inferencia de historias demográficas complejas, discernir eventos de admixtura antiguos y detectar la acción de la selección natural sobre segmentos genéticos específicos. La aplicación del modelo no se limita a humanos; el estudio exploró también otras especies como murciélagos, delfines, chimpancés y gorilas, con resultados variados que reflejan las diferencias en las historias poblacionales de cada especie. Esto demuestra la versatilidad del enfoque para explorar la dinámica evolutiva en una amplia gama de organismos. Por otro lado, el modelo tiene ciertas limitaciones inherentes al uso de supuestos simplificadores, como la asunción de un solo evento puntual de admixtura y poblaciones con tamaños efectivos constantes durante periodos clave. Además, la influencia de procesos como la selección natural, la variabilidad en tasas de mutación o recombinación, y eventos más complejos de flujo génico continuo pueden afectar los resultados.